













Die Analyse von Genprodukten in Zellen ist ein wichtiges Werkzeug für die Diagnose von Krankheiten und das Design neuer Wirkstoffe in der biologischen und medizinischen Forschung. Am Helmholtz Zentrum München wurde nun eine Methode der RNA-Sequenzierung (Transkriptomanalyse) entwickelt, die kleinste Mengen von Gentranskripten in einzelnen Zellen zielgenau erfasst. Mit dem Verfahren lassen sich einzelne ausgewählte Moleküle in einer Probe kennzeichnen und anreichern, um ihre zelluläre Funktion zu untersuchen. Damit wird es möglich, selektiv Gene in ihrer zellulären Umwelt mit hoher Genauigkeit zu charakterisieren. Die Arbeit wurde jetzt in Genome Biology veröffentlicht.
Einzelzell-RNA-Sequenzierung beruht auf der Untersuchung der molekularen Abschriften, die von aktiven Bereichen des Erbgutes in den individuellen Zellen erstellt werden. Je nach Typ und Entwicklungsstadium aktivieren Zellen unterschiedliche Gene, die von RNA-Molekülen abgelesen und in Proteine übersetzt werden. Die Anzahl der mRNA-Moleküle oder Transkripte pro Gen in einer bestimmten Zelle, kann uns über ihre Identität und ihre physiologische Reaktion auf interne oder externe Signale informieren. Das können Erkrankungen, Alterungsprozesse, Umwelteinflüsse oder Reaktionen auf pharmakologische Wirkstoffe sein. Der Nachweis von Genen allerdings, die nur in moderaten bis niedrigen Konzentrationen exprimiert werden, stellt die derzeit verwendeten Einzelzell-RNA-Sequenzierungstechniken vor große Herausforderungen. Bei der Analyse werden vorrangig so genannte Hauskeeping-Gene erkannt, die im Unterschied zu dynamisch regulierten Genen ständig exprimiert sind.
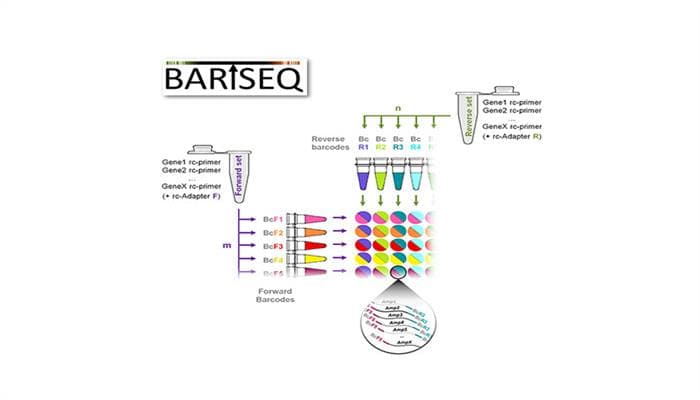
Dieses Problem umgeht die von einem Team um Dr. Micha Drukker, Institut für Stammzellforschung, und Doktorandin Fatma Uzbas entwickelte Methode BART-Seq, indem sie ausgewählte Transkripte für die Sequenzierung anreichert. BART-Seq steht für „Barcode Assembly foR Targeted Sequencing“. Dabei werden Primersätze und DNA-Barcodes kombiniert, so dass sie die Transkripte von gewünschten Genen amplifizieren können. „Wir haben eine neuartige Technik entwickelt, Primer mit DNA-Barcode durch eine einfache Synthesereaktion zu indexieren“, erläutert Micha Drukker. Sequenzierte Transkripte können auf diese Weise bis zu den einzelnen Zellen, aus denen sie stammen, zurückverfolgt werden. Da die Analyse gleichzeitig nur die ausgewählten Gene im Visier hat, ist es möglich, hochaufgelöste Informationen über diese Gene zu erhalten und so Zelle für Zelle individuell zu charakterisieren.
Das Verfahren ist preisgünstig und erfordert keine spezialisierte und teure Instrumentierung. Jede Forschergruppe mit Zugang zu Next Generation-Sequencing-Instrumenten kann BART-Seq sowohl für Einzelzellen, als auch für die Sammelanalyse von RNA oder genomischer DNA aus tausenden von Proben einsetzen.
Zusammen mit Philipp Angerer, Nikola Müller und Fabian Theis vom Institut für Computational Biology des Helmholtz Zentrums München hat Drukkers Team Software für das Design von Primern und Barcodes sowie für die Analyse der Sequenzierungsdaten entwickelt. Um die Methode für alle Forschergruppen zugänglich zu machen, ist die Software im Internet frei verfügbar.
Micha Drukker und sein Teamkolleginnen und Kollegen wünschen sich, dass ihr Verfahren ein fester Bestandteil des Toolkits der Grundlagen- und Anwendungsforschung wird. Gerade Projekte zum Wirkstoffscreening, wie die Messung der Reaktion von kultivierten β-Zellen auf Medikamente, oder beispielsweise die zielgenaue Bearbeitung von Genomsequenzen mit CRISPR-Cas9 könnten von BART-Seq stark profitieren.